


In this article, Debarati Tripathi who is currently pursuing M.A. IN BUSINESS LAWS, from NUJS, Kolkata, discusses Indian laws and policy on generic drugs.
Introduction
- The Pharmaceutical industry in India was more or less non-existent prior to 1947, there were no production units of allopathic medicine in the country. Today, in the 21st century the Indian pharma sector is well recognized and a major provider of medicines and healthcare products globally.*India is now a leading pharmaceutical producer, with a fast-growing generics and biosimilar market. India currently ranks fourth in the world among the highest generic pharmaceuticals producers and contributes 20% of global generic drug exports – as per a report by Equity Master.
- Several attractive reasons for India’s rising prominence in pharmaceuticals can be cited, including economic labor, strong government support, infrastructure and legislature, and lower production costs. While India’s domestically-owned pharmaceuticals companies may be few in number, many multinational pharma giants appear to be taking advantage of the country’s inexpensive labor through India-based subsidiaries. India also boasts lower research and development (R&D) and manufacturing costs, given government initiatives that support the pharmaceuticals sector, including fiscal incentives and streamlined development procedures.
- The cost of production has been a leading source of India’s industry strength, as India is 60% cheaper than the U.S. and 50% cheaper than Europe in terms of drug production costs.
- The growth and development of any business are dependent on infrastructure, policies, and legislations of the land for ease of doing business; Pharma is no different. It is worthwhile to mention that the flexible provisions of the Patent Act of 1970 and other supportive policies of the Government of India played an instrumental role in the growth and development of this industry.
- We recognize the importance of public policies in influencing the present structure of the industry and aim to present in brief the important policy changes that have taken place in this sector and also the current status of the legislature.
In India, the approval, production, and marketing of quality drugs at reasonable prices is ensured by the following regulatory bodies,
The Central Drug Standards and Control Organization (CDSCO),
- CDSCO functions under the Ministry of Health and Family Welfare
- Prescribes standards and measures for ensuring the safety, efficacy, and quality of drugs, cosmetics, diagnostics and devices in the country;
- Regulates the market authorization of new drugs and clinical trials standards;
- Supervises drug imports and approves licenses to manufacture the above-mentioned products;
The National Pharmaceutical Pricing Authority (NPPA), 1997
- The NPPA functions under the Department of Chemicals and Petrochemicals.
- fixes or revises the prices of decontrolled bulk drugs and formulations periodically
- updates the list under price control through inclusion and exclusion of drugs in line with prescribed guidelines priodically;
- maintains data on production, exports and imports and market share of pharmaceutical firms;
- monitors the shortage of medicines in addition to providing inputs to Parliament in issues pertaining to drug pricing.
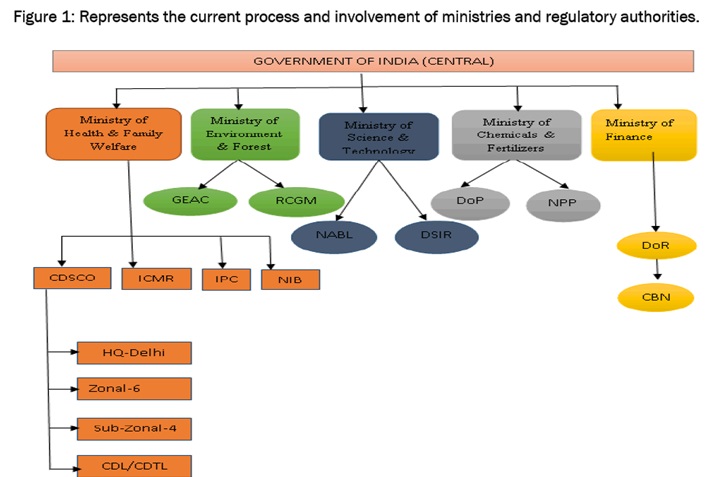
The Ministry of Health and Family Welfare examines pharmaceutical issues within the larger context of public health while the focus of the Ministry of Chemicals and Fertilizers is on industrial policy. In July 2008,the cabinet Secretariat, created a new department under Ministry of Chemicals and Fertilisers – the Department of Pharmaceuticals, with the objective of giving greater focus and thrust on the development of Pharmaceutical Sector in India and to regulate various complex issues related to pricing and availability of affordable medicines, research & development, protection of intellectual property rights and international commitments related to pharmaceutical sector which require integration of work with other ministries.
All the drugs and pharmaceuticals, unless specifically allotted to any other department, would come under the purview of the Department of Pharmaceuticals. The main functions and responsibilities of the Department are as follows,
- All matters relating to NPPA including its functions of price control and monitoring.
- Responsible for the drugs and pharmaceuticals, excluding those specifically allotted to other departments, and for the development of infrastructure, manpower and skills for the pharmaceutical sector
- Work for the promotion and coordination of basic, applied and other research in areas related to the pharmaceutical sector and for international co-operation in pharmaceutical research.
- Entrusted with the task of maintaining inter-sectoral coordination between organizations and institutes, both under Central and State Governments, related to areas concerning the subject.
- To deal with all matters relating to planning, development, and control of, and assistance to, all industries in the pharmaceutical segment.
- Promotion of Public Private Partnership (PPP) in pharmaceutical related areas.
However, other ministries also play a role in the regulation process.
- The Ministry of Environment and Forests, Ministry of Finance, Ministry of Commerce and Industry and the Ministry of Science and Technology also have a part to play in the regulation process. The process for drug approval requires the coordination of different departments, in addition to theDCGI, depending on whether the application in question is a biological drug or one based on recombinant DNA technology.
- The Department of Industrial Policy and Promotion and Directorate General of Foreign Trade, both under the aegis of Ministry of Commerce and Industry and the Ministry of Chemicals and Fertilizers, look into matters related to industrial policy such as the regulation of patents, drug exports, and government support to the industry.
- Licensing, quality control issues, market authorization is regulated by the Central Drug Controller, Ministry of Health and Family Welfare, Department of Biotechnology, Ministry of Science and Technology (DST) and Department of Environment, Ministry of Environment and Forests.
State drug controllers have the authority to issue licenses for the manufacture of approved drugs and monitor quality control, along with the Central Drug Standards Control Organization (CDSCO).
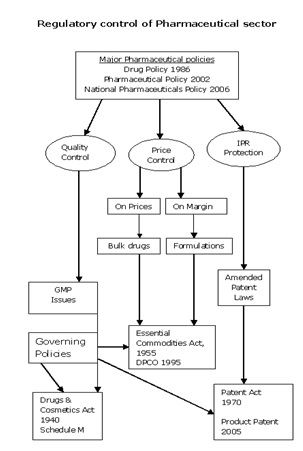

The important regulatory mechanisms are summarized below to give an understanding of the various legislatures that are present and govern the Pharmaceutical practice in India.
Drugs and Cosmetics Act of 1940 and Rules 1945
In India, drug manufacturing, quality and marketing is regulated in accordance with the Drugs and Cosmetics Act of 1940 and Rules 1945. Over the last few decades, this act has undergone several amendments. The Drugs Controller General of India (DCGI), who heads the Central Drugs Standards Control Organization (CDSCO), assumes responsibility for the amendments to the Acts and Rules. Other major related Acts and Rules include the Pharmacy Act of 1948, The Drugs and Magic Remedies Act of 1954 and Drug Prices Control Order (DPCO) 1995 and various other policies instituted by the Department of Chemicals and Petrochemicals.
Some of the important schedules of the Drugs and Cosmetic Acts include:
- Schedule D: dealing with exemption in drug imports,
- Schedule M: to control spurious drugs, incorporated in 1995 that lays down Good Manufacturing Practices(GMP) at par with WHO standards.involving premises and plants
- Schedule Y: which, specifies guidelines for clinical trials, import and manufacture of new drugs
In accordance with the Act of 1940, there exists a system of dual regulatory control or control at both Central and State government levels. The central regulatory authority undertakes approval of new drugs, clinical trials, standards setting, control over imported drugs and coordination of state bodies’ activities. State authorities assume responsibility for issuing licenses and monitoring manufacture, distribution and sale of drugs and other related products.
The Act has been amended several times as listed below,
- The Drugs (Amendment) Act, 1955 (11 of 1955)
- The Drugs (Amendment) Act, 1960 (35 of 1960)
- The Drugs (Amendment) Act, 1962 (21 of 1962)
- The Drugs and Cosmetics (Amendment) Act, 1964 (13 0f 1964)
- The Drugs and Cosmetics (Amendment) Act, 1955 19 of 1972)
- The Drugs and Cosmetics (Amendment) Act, 1982 (68 of 1982)
- The Drugs and Cosmetics (Amendment) Act, 1986
- The Drugs and Cosmetics (Amendment) Act, 1995 (71 of 1995)
Narcotic Drugs And Psychotropic Substances Act
The Narcotic Drugs and Psychotropic Substances Bill, 1985 was introduced in the Lok Sabha in August 1985 and subsequently passed by both the Houses of Parliament.It came into force on 14 November 1985 as The Narcotic Drugs And Psychotropic Substances Act, 1985 (shortened to NDPS Act). Under the NDPS Act, it is illegal for a person to produce/manufacture/cultivate, possess, sell, purchase, transport, store, and/or consume any narcotic drug or psychotropic substance. Under one of the provisions of the act, the Narcotics Control Bureau was set up with effect from March 1986. The Act is designed to fulfill India’s treaty obligations under the Single Convention on Narcotic Drugs, Convention on Psychotropic Substances, and United Nations Convention Against Illicit Traffic in Narcotic Drugs and Psychotropic Substances. The Act has been amended three times – in 1988, 2001, and most recently in 2014.
Prevention of Illicit Trafficking in Narcotic Drugs and Psychotropic Substances Act
- The Prevention of Illicit Trafficking in Narcotic Drugs and Psychotropic Substances Act is a drug control law passed in 1988 by the Parliament of India. It was established to enable the full implementation and enforcement of the Narcotic Drugs and Psychotropic Substances Act of 1985.
- The Narcotics Control Bureau (NCB) is the chief law enforcement and intelligence agency of India responsible for fighting drug trafficking and the abuse of illegal substances. It was created on 17 March 1986 to enable the full implementation of the Narcotic Drugs and Psychotropic Substances Act (1985) and fight its violation through the Prevention of Illicit Trafficking in Narcotic Drugs and Psychotropic Substances Act (1988).
- There exists a published list that mentions the names of all substances banned or controlled in India under the NDPS Act – 237 line items. The list uses the International Nonproprietary Name (INN) of the drugs but in some cases mentions drugs by their chemical name also widely known drugs such as ganja, cocaine, heroin etc. are mentioned as such. Cultivation/production/manufacture, possession, sale, purchase, transport, storage, consumption or distribution of any of the following substances, except for medical and scientific purposes and as per the rules or orders and conditions of licenses that may be issued, is illegal and a punishable offense.
The Patents Act
- Concerned by the high price of medicines and the lack of domestic infrastructure, the government constituted the Hathi Committee in 1974 ‘to probe into the problems and suggest a rational drug policy that would meet the medicinal needs of the country’. Recommended by the Committee’s report, the government amended the Patent Act of 1970 and enacted the Foreign Exchange Regulation Act (FERA) 1973 in its New Drug Policy (NDP) of 1978.
- The Patent Act of 1970 recognized only process patents. The life of the patent was also reduced significantly from 16 to 5 years from the date of sealing or 7 years from the date of filling a complete application, whichever is shorter; in other words, the maximum period of patent was 7 years. Further, in the amended Act an MNC could patent only one process.
- The Patent Act of 1970 and the changes in domestic regulation virtually curbed the monopoly of MNCs. Adopting the flexible provisions of the amended patent act, indigenous companies started imitating the patented product and could eventually come out with better processes for the same product.
- The industry also embarked on the path of high growth during this period. The other significant outcomes were fall in the prices of the medicines and the introduction of a large number of generic versions of patented products. The drug policy of 1978 was, however, revised in 1986 to dilute the mechanism of check and control with respect to the production of certain categories of drugs. NDP 1986 also regularized the production of a large number of drugs that were earlier questionable on regulatory grounds.
- The Patent Law was amended under the WTO compulsion to recognize product patent from 2005 onward. This was implemented in a staggered manner in three phases. The first phase of it was implemented in 1995 in which the ‘mail-box’ system was recognized.
- On January 1, 2000, a Second Amendment was introduced where the salient features were re-defined patentable subject matter, extended the term of patent protection to 20 years and amended the compulsory licensing system.
- A third amendment of patent law was made on January 1, 2005 to introduce product patent regime in areas, including pharmaceuticals that were hitherto covered by process patents only.
In summary, there is a gradual shift in public policy from the regime of control and process patents to a regime of decontrol and product patents.
Pricing and tax policies through DPCO and NPPA
- Price control on medicines was first introduced in India in 1962 and has subsequently undergone evolution through the Drug Price Control Order (DPCO). As per the directive of NPPA, the criterion for price regulation is based on the nature of the drug in terms of whether it enjoys mass consumption and in terms of whether there is lack of adequate competition for the drug.
- In 1978 selective price controls based on disease burden and prevalence was brought about by the Government. Thereafter, the list of prices under DPCO underwent a gradual decrease over a period of time. Around 80% of the market, with 342 drugs, was under price control in 1979. The number of drugs under DPCO decreased from 142 drugs in 1987 to 74 in 1995.
- The major objective of DPCO 1995 was to decrease monopoly in any given market segment, further decrease the number of drugs under price control to 74 and the inclusion of products manufactured by small scale producers under price control list.
In 1997, the National Pharmaceutical Pricing Authority [NPPA] was constituted in order to administer DPCO and deal with issues related to price revision.
- The NPPA also regulates the prices of bulk drugs or pharmaceutical actives. The MRP excise on medicines was levied by the Finance ministry in 2005 with the objective of increasing revenue and lowering prices of medicines by using fiscal deterrent on MRP. This change may have had some impact in terms of magnifying the advantage to industries located in the excise free zones.
- Drugs with high sales and a market share of more than 50% are part of the price regulation exercise. These drugs are referred to as scheduled drugs. Historically the NPPA would intervene only if the annual price increases were more than 20%.
- However, post-2007, the NPPA intervenes in cases where drugs have significant sales and where the annual price increases by 10%.
- The National Pharmaceuticals Policy 2006, proposed various measures such as increasing the number of bulk drugs under regulation from 74 to 354, regulating trade margins and instituting a new framework for drug price negotiations so as to make drugs more affordable for the Indian masses, to name a few.
Good Manufacturing Practices and policies
- World Health Organization GMP guidelines were instituted in 1975 in order to assist regulatory authorities in different countries to ensure consistency in quality, safety and efficacy standards while importing and exporting drugs and related products. India is one of the signatories to the certification scheme.
- The WHO-GMP certification, which possesses two-year validity, may be granted both by CDSCO and state regulatory authorities after a thorough inspection of the manufacturing premises.
- WHO defines Good manufacturing practice (GMP) as a system for ensuring that products are consistently produced and controlled according to quality standards. It is designed to minimize the risks involved in any pharmaceutical production be it manufacturing, packaging, testing, labeling, distributing and importing, that cannot be eliminated through testing the final product – drug or device or any formulation. The GMP protocols are largely concerned with parameters such as drug quality, safety, efficacy and potency.
- India has made progress in the domain of GMP through the enforcement of Schedule M Compliance. The requirements specified under the upgraded Schedule ‘M’ for GMP have become mandatory for pharmaceutical units in India from July 1, 2005. Schedule M classifies the various statutory requirements mandatory for drugs, medical devices and other categories of products as per the current Good Manufacturing Practices (cGMP). Schedule M contains various regulations for manufacturing, premises, waste disposal, and equipment.
- Schedule M protocols have been revised to harmonize it along the lines of WHO and US-FDA protocols. These revised protocols include detailed specifications on infrastructure and premises, environmental safety and health measures, production and operation controls, quality control and assurance and stability and validation studies.
- Schedule M compliance is the next thing, smaller pharmaceutical units may take longer to be compliant whereas large-scale firms have shown greater willingness to comply with the revised norms in order to increase their competitiveness in the global arena. According to state regulatory sources, units in states like Gujarat, Karnataka, Maharashtra and Andhra Pradesh have achieved a high percentage of Schedule M compliance in comparison to units in other states.
- Export of drugs, devices, and formulations to developed countries from India requires that Regulators from these countries visit Indian manufacturers to carry out a thorough inspection of their manufacturing units before registering the concerned product.
- A large number of domestic players are seeking international regulatory approvals from agencies like US-FDA, MHRA UK, TGA Australia and MCC South Africa in order to export their products, mostly generic medicines, in these markets. Indian drug makers have the largest number of FDA-approved plants outside the US and accounted for 39 per cent of all approvals for generic drugs during 2013.
Policies relating to clinical trials
- Till about a decade ago, there was little or no visibility with regard to the conduct of quality clinical trials in India-compliant to regulatory standards and ethics. The Central Drugs Standards Control Organization (CDSCO) has played a critical role in bringing about a positive change in the clinical trials landscape for India.The progression towards Good Clinical Practice (GCP) has largely been a gradual and slow process.
- In 1988 local clinical trials for new drug introductions were first made mandatory in India. Along with the changeover to product patents in January 2005, India also amended the schedule Y of the ‘‘Drugs and Cosmetics Rules’’ to allow drug trials without a phase lag in the country, that is, Phase II and Phase III trials were permitted only after these had been carried out elsewhere in the world.
- The new rule permitted conducting the concurrent trials of the same phase in India. Also Drugs Technical Advisory Board (DTAB) made GLP practices mandatory for all laboratories and in-house units of pharmaceutical firms and Contract Research Organizations (CROs).
- Reports of incidents of ethical violations related to informed consent and conduct of trials by multinational and domestic organizations were known prior to the year 2000. In 2000, the regulators – the Central Ethics Committee on Human Research (CECHR) and Indian Council of Medical Research (ICMR) took proactive initiative to conceptualize and issue the Ethical Guidelines for Biomedical Research on Human Subjects.
- The Good Clinical Practice (GCP) guidelines were developed in line with the latest WHO and ICH guidelines in 2001 by Central Expert Committee -set up for the purpose by Central Drugs Standards Control Organization (CDSCO)
- Subsequently in 2005, the requirements of data submission on animal testing for permission to undertake Phase I, Phase II and Phase III clinical trials were laid down in the revised Schedule Y of the Drugs and Cosmetics rules.
- Clear responsibilities for investigators; and sponsors were specified and notifying changes in the protocol were made mandatory. Expert clinicians & scientists from the industry assist the evaluation of the relevant data submitted to the Drugs Control General of India (DCGI)
- Similarly, for registration and approval of new drugs, which have already been registered and used in the country of origin, The DCGI mandates Phase II trials in about 100 prior to allowing such products to be marketed in India. Normally, new drug approval is usually granted for a period of about two years. The trials are conducted only after clearances are obtained from the Institutional Ethics Committees. Consent of patients for participation in such trials is an integral part of the regulatory framework.
- However, there remains a need for the establishment of pharmacovigilance centers at national, zonal and regional levels to monitor adverse drug reactions to be met.
Policies and guidelines are evolving in the allied fields of Medical devices and Biotechnology in India.

Medical devices
- The JJ Hospital controversy, involving the use of unapproved and untested stents on 60 patients and the subsequent recommendations made by the Mashelkar Committee in 2004 resulted in the Department of Medical Education and Research (DMER) banning the use of unapproved stents and stressing on regulatory approvals from the country of manufacture or US-FDA approval for medical devices.
- Following on from the incident, in June 2007, the DCGI introduced a new set of guidelines for the import and manufacture of medical devices in the country.
- The Mashelkar Committee subsequently recommended the creation of a specific medical devices division within the CDSCO in order to address the management, approval, certification and quality assurance of all medical devices.
- This essentially consisted in alteration of the status of sterile medical devices, intended for internal or external use to medical drugs and creation of suitable provisions and amendments to the Drugs and Cosmetics Act of 1940.
- The Drugs Consultative Committee approved these recommendations in 2005, ensuring that in future all devices would be licensed for manufacture, distributed and sold by the CDSCO, with special evaluation committees in order to ensure that the concerned manufacturing units complied with the requisite GMP requirements.
Biotechnology and related products
- The Department of Biotechnology [DBT] constituted under the Ministry of Science and Technology is the parent body for policy, promotion of R&D, international cooperation and manufacturing activities.
- Together with DBT, Genetic Engineering and Approval Committee [GEAC] constituted under Ministry of Environment and Forests [MoEF] is the key regulatory body in Biotechnology in India. Several committees have also been constituted under the said ministries to regulate the activities involving handling, manufacture, storage, testing, and release of genetic modified materials in India.
- The Institutional BioSafety Committee (IBSC), Review Committee on Genetic Manipulation (RCGM) and the Genetic Engineering Approval Committee (GEAC) to monitor rDNA research, product development and commercialization. The ISBC functions as the nodal point for interaction within the institution for the implementation of the rDNA Biosafety guidelines. The RCGM essentially monitors the safety related aspects of activities involving genetically engineering organisms or hazardous microorganisms.
- The GEAC undertakes the responsibility of approval of activities involving large-scale use of genetically modified/hazardous microorganisms and products thereof in research and industrial production and their safety in terms of environmental protection. In addition, the DCGI and state drug controllers as per the Drugs and Cosmetics Act 1945 and its subsequent amendments regulate biologicals.
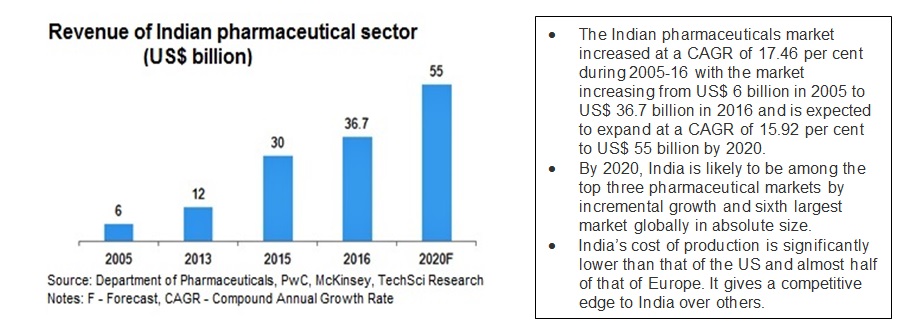
With continuous improvements in laws and policies along with mechanisms for enforcement viz. checks, audits and enablement of the sector by the Government, the pharma industry has emerged as a stable, long term sector across decades and through various economic trends. This latest report from India Brand equity, cited below shows the increasing investments in the sector, with a projection of crossing $ value of 50$ by 2020.
Bibliography :
http://www.nistads.res.in/indiasnt2008/t4industry/t4ind18.htm
Adapted from Dun & Bradstreet (D&B) 2007
https://en.wikipedia.org/wiki/Drugs_and_Cosmetics_Act,_1940
http://www.ibef.org/industry/indian-pharmaceuticals-industry-analysis-presentation
https://en.wikipedia.org/wiki/Drug_policy_of_India
 Serato DJ Crack 2025Serato DJ PRO Crack
Serato DJ Crack 2025Serato DJ PRO Crack

")


 Allow notifications
Allow notifications




